宁波特殊样本荧光定量PCR原理及步骤
PCR的反应条件:dNTP浓度过高会加快反应速度,但同时还可以增加碱基的错误掺入率。引物浓度过高会引起错配和非特异性产物扩增。TaqDNA聚合酶浓度过高会引起错配和非特异性产物扩增,低则合成产物量减少。TaqDNA聚合酶无校正功能,掺入错误率达2*E-4个核苷酸,一个30个循环的扩增反应0.1%-0.25%总错误率。在90~95度下可使整个基因组的DNA变性为单链。一般94~95度30~60s。时间过长使TaqDNA聚合酶失活和dNTP破坏增多。DNA很快冷却到40~60度使引物和模板结合。引物长度在15~25时退火温度。逆转录-聚合酶链反应较广用于表达谱,以确定基因的表达或鉴定RNA转录物的序列。宁波特殊样本荧光定量PCR原理及步骤

聚合酶链式反应的常见问题:靶序列变异:如靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某 段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。靶序列太短或引物太短,容易出现假阳性。需重新设计引物。出现片状拖带或涂抹带:PCR扩增有时出现涂抹带或片状带或地毯样带。其原因往往由于酶量过多或酶的质量差,dNTP浓度过高,Mg2+浓度过高,退火温度过低,循环次数过多引起。其对策有:减少酶量,或调换另一来源的酶。②减少dNTP的浓度。适当降低Mg2+浓 度。增加模板量,减少循环次数。黄山分子生物学PCR检测技术聚合酶链式反应中要确保反应缓冲液融化完全并彻底混匀。
聚合酶链反应注意事项:在实验过程中要防止RNA的降解,保持RNA的完整性。在总RNA的提取过程中,注意避免mRNA的断裂;为了防止非特异性扩增,必须设阴性对照;内参的设定:主要为了用于靶RNA的定量。常用的内参有G3PD(甘油醛-3-磷酸脱氢酶)、β-Actin(β-肌动蛋白)等。其目的在于避免RNA定量误差、加样误差以及各PCR反应体系中扩增效率不均一各孔间的温度差等所造成的误差;PCR不能进入平台期,出现平台效应与所扩增的目的基因的长度、序列、二级结构以及目标DNA起始的数量有关。故对于每一个目标序列出现平台效应的循环数,均应通过单独实验来确定;防止DNA的污染: 采用DNA酶处理RNA; 在可能的情况下,将PCR引物置于基因的不同外显子,以消除基因和mRNA的共线性。
聚合酶链式反应的常见问题:出现非特异性扩增带:PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带 与非特异性扩增带。非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。其次是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。其对策有:必要时重新设计引 物。减低酶量或调换另一来源的酶。降低引物量,适当增加模板量,减少循环次数。适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸)。聚合酶链反应可以用于分析病症、微生物或其他疾病状态中基因表达水平的变化。
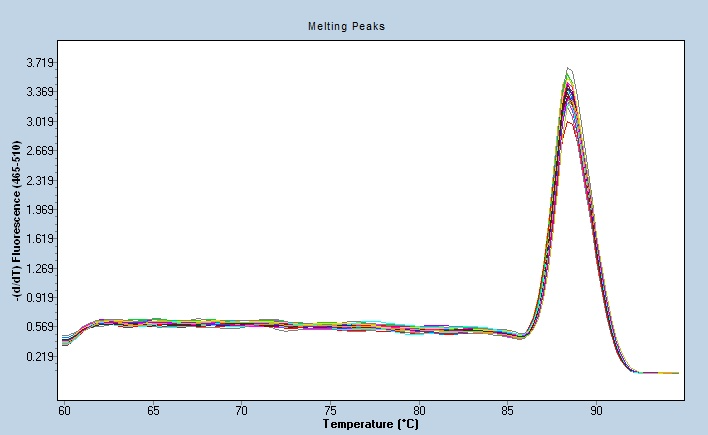
聚合酶链式反应的循环参数:预变性,模板DNA完全变性与PCR酶的完全对PCR能否成功至关重要,建议加热时间参考试剂说明书,一般未修饰的Taq酶时间为两分钟。变性步骤,循环中一般95℃,30秒足以使各种靶DNA序列完全变性,可能的情况下可缩短该步骤时间。变性时间过长损害酶活性,过短靶序列变性不彻底,易造成扩增失败。引物退火,退火温度需要从多方面去决定,一般根据引物的Tm值为参考,根据扩增的长度适当下调作为退火温度。然后在此次实验基础上做出预估。退火温度对PCR的特异性有较大影响。引物延伸,引物延伸一般在72℃进行(Taq酶很适温度)。但在扩增长度较短且退火温度较高时,本步骤可省略延伸时间随扩增片段长短而定,一般推荐在1000bp以上,含Pfu及其衍生物的衍生设定为1min/kbp。循环数,大多数PCR含25-35循环,过多易产生非特异扩增。延伸,在一个循环后,反应在72℃维持10-30分钟.使引物延伸完全,并使单链产物退火成双链。PCR的很大特点是能将微量的DNA大幅增加。武汉血液荧光定量PCR应用
逆转录聚合酶链反应(逆转录-聚合酶链反应):用于从RNA中扩增DNA。宁波特殊样本荧光定量PCR原理及步骤
聚合酶链反应的常见问题分析与解决方法:反应缓冲液未完全融化或未充分混匀。确保反应缓冲液融化完全并彻底混匀。引物特异性差。利用BLAST检查引物特异性或重新设计引物。引物量过多。减少反应体系中引物的用量。模板量过多。质粒DNA的用量应<50ng,而基因组DNA则应<200ng。外源DNA污染。确保操作的洁净。阴性对照出现条带:试剂,头,工作台污染。使用全新的试剂和头,对工作台进行清洁。条带大小与理论不符:污染。使用全新的试剂和头,对工作台进行清洁。模板或引物使用错误。更换引物和模板。基因亚型。对研究的基因进行序列分析和BLAST研究。宁波特殊样本荧光定量PCR原理及步骤
上一篇: 南京微量PCR检测技术
下一篇: 无锡血液荧光定量PCR设计公司